Computational, evolutionary and human genomics at Stanford


- Event Report
- Fellows Features
- Modeling & Theory in Population Biology
- Press
- Research Summaries
- Uncategorized
This is the blog for the Center for Computational, Evolutionary, and Human Genomics at Stanford.
Latest Posts:
-
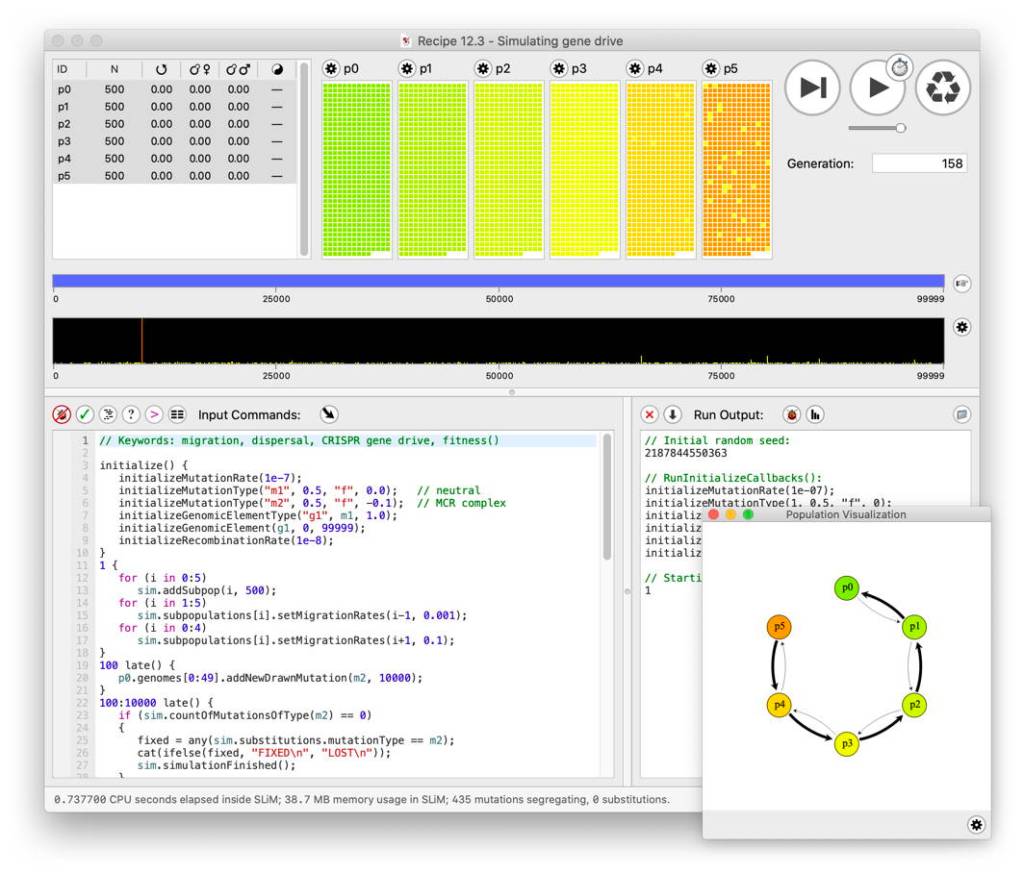
Modeling & Theory in Population Biology: Software Series – Ben Haller – SLiM
For the second installment of MTPB’s software series, Ben Haller of Cornell University spoke about SLiM on March 13, 2024. Ben began by acknowledging the main contributors to the project besides himself. Philip Messer wrote the original SLiM, and Peter Ralph is a major contributor – however, Ben acknowledged that many others have also contributed… Read more
-

Fellows Feature: Nandita Garud
Can you tell us about your research? What were you working on as a fellow, and how has it progressed since completing the fellowship? My research in Dr. Dmitri Petrov’s lab as a graduate student was on understanding the rapidity of adaptation in natural populations of Drosophila melanogaster. Adaptation is a process whereby a population… Read more
-

Modeling & Theory in Population Biology Session 6 – Mia Miyagi, Francois Bienvenu
On February 28, 2024, Francois Bienvenu and Mia Miyagi presented on models that have been relevant to their respective research areas. Francois Bienvenu from Laboratoire de Mathématiques de Besançon began with his presentation: “Where do phylogenetic trees come from?” Starting by drawing a basic phylogenetic tree, Bienvenu noted that it is a good structure to… Read more
-
Modeling & Theory in Population Biology Session 5 – MontGomery Slatkin, Sally Otto, John Wakeley
On February 20, 2024, John Wakeley and Sally Otto spoke with Montgomery Slatkin, Professor Emeritus in the Department of Integrative Biology at the University of California, Berkeley, on how the field of modeling has developed over the years. The following is a summary of their conversation, minimally paraphrased and edited for clarity. Otto: When was… Read more
-

Modeling & Theory in Population Biology Session 3 – Ben Ashby, Caroline Colijin, Laura Kubatko, Daniel Weissman
On January 30, 2024, four panelists from three institutions sat down with Ailene MacPherson to talk about the unique paths of their careers as interdisciplinary researchers. The following is a summary of their conversation, paraphrased and edited for clarity. Give a brief description of your career path, and how you ended up in your department.… Read more
-

Modeling & Theory in Population Biology Session 2 – Bruce Weir
For the second installment of the Modeling & Theory in Population Biology series on January 17, 2024, Bruce Weir presented his talk on applications for population genetics in forensics. He opened with a recent homicide case as an example, in which DNA from the involved weapon’s trigger matched the DNA profile of a person of… Read more
-

Modeling & Theory in Population Biology Session 1 – Maria Servedio, Marc Feldman, Joel E. Cohen, Tanja Stadler
The January 11, 2024 kickoff event of the Modeling & Theory in Population Biology series featured four speakers: Maria Servedio from the University of North Carolina, Marc Feldman from Stanford, Joel E. Cohen from Rockefeller University, and Tanja Stadler from ETH Zürich. This group of speakers reflected the initial inspiration for this program: how diverse… Read more
-

Fellows Feature: Nicolas Alcala
Nicolas Alcala was a 2014-15 fellow who has gone on to become a computational biologist for the World Health Organization, focusing on multi-omic integration and cancer evolution for the Rare Cancers Genomics Initiative. How did you first get involved with the center? What was your original focus as a fellow, and how did it evolve… Read more
-

Fellows Feature: Clare Abreu
Clare’s fellowship in 2020 was a crucial step in her unusual career journey from journalist to biologist. Read on to learn about the inspiration behind her research! Can you tell us about your research? What were you working on as a fellow, and how has it progressed since completing the fellowship? I study evolution in… Read more
-

FELLOWS FEATURE: ALANA PAPULA
Alana Papula is a member of our most recent cohort of CEHG fellows, studying evolution of bacterial populations in the Fisher lab. Read on for a peek into her experience during the fellowship! Can you tell us about your research? What were you working on as a fellow, and how has it progressed since completing… Read more
